New machine-learning algorithms may revolutionize drug discovery — and our understanding of life
February 8, 2017
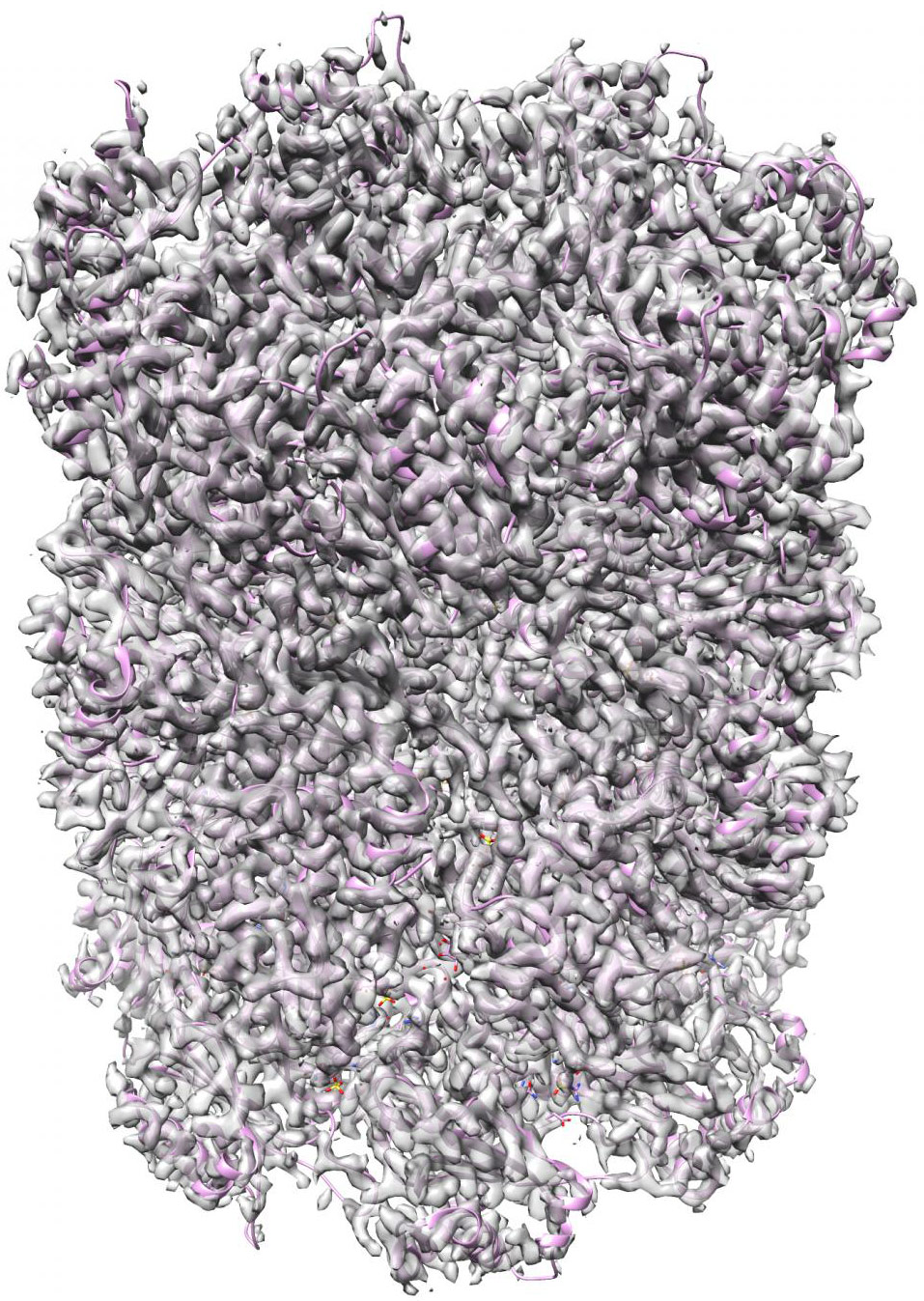
A new set of machine-learning algorithms can generate 3D structures of complex nanoscale protein molecules like this complex proteasome map refined to 2.8 Angstroms (.28 nanometer) in 70 min with 49,954 particle images (credit: Structura Biotechnology Inc.)
A new set of machine-learning algorithms developed by researchers at the University of Toronto Scarborough can generate 3D structures of nanoscale protein molecules that could not be achieved in the past. The algorithms may revolutionize the development of new drug therapies for a range of diseases and may even lead to better understand how life works at the atomic level, the researchers say.
Drugs work by binding to a specific protein molecule and changing the protein’s 3D shape, which alters the way the drug works once inside the body. The ideal drug is designed in a shape that will only bind to a specific protein or group of proteins that are involved in a disease, while eliminating side effects that occur when drugs bind to other proteins in the body.
A significant computational problem
Since proteins are tiny — about 1 to 100 nanometers — even smaller than the shortest wavelength of visible light, they can’t be seen directly without using sophisticated techniques like electron cryomicroscopy (cryo-EM). Cryo-EM uses high-power microscopes to take tens of thousands of low-resolution images of a frozen protein sample from different positions.
The computational problem is to then piece together the correct high-resolution 3D structure from these 2D images.
Existing techniques take several days or even weeks to generate a 3D structure on a cluster of computers, requiring as much as 500,000 CPU hours, according to the researchers. Also, existing techniques often generate incorrect structures unless an expert user provides an accurate guess of the molecule being studied.

CryoSPARC machine learning algorithms can generate 3-D structures of nanoscale protein molecules (credit: Structura Biotechnology Inc)
New high-speed, deep-learning algorithms
That’s where the new set of algorithms* comes in. It reconstructs 3D structures of protein molecules using these images. “Our approach solves some of the major problems in terms of speed and number of structures you can determine,” says Professor David Fleet, chair of the Computer and Mathematical Sciences Department at U of Toronto Scarborough.
The algorithms could significantly aid in the development of new drugs because they provide a faster, more efficient means at arriving at the correct protein structure.
The new approach, called cryoSPARC, developed by the team’s startup, Structura Biotechnology Inc., eliminates the need for that prior knowledge and can make the computations possible in minutes on a single computer, using a standalone graphics processing unit (GPU) accelerated software package, according to the researchers.
The research was published in the current edition of the journal Nature Methods. It received funding from the Natural Sciences and Engineering Research Council of Canada (NSERC). The new cryo-EM platform is already being used in labs across North America, the researchers note.
* “We use an SGD [stochastic gradient descent] optimization scheme to quickly identify one or several low-resolution 3D structures that are consistent with a set of observed images. This algorithm allows for ab initio heterogeneous structure determination with no prior model of the molecule’s structure. Once approximate structures are determined, a branch-and-bound algorithm for image alignment helps rapidly refine structures to high resolution. The speed and robustness of these approaches allow structure determination in a matter of minutes or hours on a single inexpensive desktop workstation. … SGD was popularized as a key tool in deep learning for the optimization of nonconvex functions, and it results in near human-level performance in tasks like image and speech recognition.” — Ali Punjani et al./Nature Methods
University of Toronto Scarborough | New algorithms may revolutionize drug discoveries and our understanding of life
Abstract of cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination
Single-particle electron cryomicroscopy (cryo-EM) is a powerful method for determining the structures of biological macromolecules. With automated microscopes, cryo-EM data can often be obtained in a few days. However, processing cryo-EM image data to reveal heterogeneity in the protein structure and to refine 3D maps to high resolution frequently becomes a severe bottleneck, requiring expert intervention, prior structural knowledge, and weeks of calculations on expensive computer clusters. Here we show that stochastic gradient descent (SGD) and branch-and-bound maximum likelihood optimization algorithms permit the major steps in cryo-EM structure determination to be performed in hours or minutes on an inexpensive desktop computer. Furthermore, SGD with Bayesian marginalization allows ab initio 3D classification, enabling automated analysis and discovery of unexpected structures without bias from a reference map. These algorithms are combined in a user-friendly computer program named cryoSPARC